IVDR Report Updates 2025
As we approach the close of 2025, the IVDR regulatory landscape continues to take shape. Throughout the year, we’ve been sharing regular updates on the changes ahead, and this latest update brings some genuinely encouraging signs of progress worth highlighting.
IVDR End of Year Round-up: December 2025
EUDAMED
Perhaps the most important announcement for this update is that the EU Commission have, finally, announced a date for partial implementation of the devices database EUDAMED. Following the announcement on 25th November 2025, use of the first 4 modules of EUDAMED will become mandatory on 28th May 2026.These modules are:
- Actor registration
Enables registration of economic operators and provides Actor ID/Single registration number (SRN) Actor registration module - Public Health - European Commission - UDI/Devices registration
Enables registration of unique device identifiers (UDI) UDI/Device registration - Public Health - European Commission - Notified Bodies & Certificates
Gives data provided by Notified Bodies (NB) on certificates of conformity Notified Bodies and Certificates module - Public Health - Market Surveillance
Primarily aimed at providing information from Competent Authorities (CA) 875ff13b-7680-4bca-b0f8-c7aec95e565b_en
Revision of the Regulations and Call for Feedback
The EU Commission published an amending act for MDR and IVDR in recognition of the problems with the regulations, putting forward a large number of amendments. The document itself is an eye-watering 170 pages but there’s a good summary from B.S. Partnership here: EU MDR & IVDR Amendment 2025: Key Changes for Manufacturers — BS Partnership
The EU Commission then sought feedback on the proposed changes with an initiative entitled: “Targeted revision of the EU rules for medical devices and in vitro diagnostics” (see Medical devices and in vitro diagnostics – targeted revision of EU rules for full details).
The Commission recognised the “challenges” associated with implementation both of MDR and IVDR and has faced numerous requests from the EU parliament, individual member states and various stakeholders to simplify the regulatory framework.It acknowledged that the overly onerous regulatory environment imposed by the regulations could potentially lead to the withdrawal of devices from the EU possibly affecting patient health and could also reduce the competitiveness of the EU as a marketplace for medical devices and IVDs, disproportionately affecting SMEs.Therefore, the Commission initiated a targeted revision strategy which aims: “to simplify and streamline the regulatory framework, and make it more cost-efficient and proportionate, while preserving a high level of public health and patient safety and maintaining the overall structure of the current regulatory framework”.It then ran a consultation exercise to collect feedback from stakeholders, although not the public, about proposed revisions directed particularly towards SMEs.
The aims of the proposed revisions, given in the amendments act and call for evidence were:
- reduce the administrative burden including reporting obligations;
- enhance the predictability and cost-efficiency of the certification processes of notified bodies;
- make the conformity assessment requirements more proportionate, especially for low- and medium- risk devices and those that cater to special patient needs;
- enable further digitalisation;
- streamline procedures including those on governance;
- enable the EU medical device sector to benefit from international cooperation including reliance, where appropriate;
- better align the regulatory framework with other relevant legislation.
The consultation ran from 8th September 2025 – 6th October 2025 and the results are now published on Europa (Medical devices and in vitro diagnostics – targeted revision of EU rules).
There were 441 responses received with nearly a quarter from Germany alone and approximately 62% from businesses or business associations. Of the 441, 173 were specifically focused on IVDR (i.e. filtered out using “IVDR” as the keyword).
No breakdown of the data specific to IVDR was given and Diaceutics will, therefore, publish a breakdown and sentiment analysis of IVDR-specific responses early in the New Year.
Notified Bodies (NB)
There are now 19 NB with a third one in Finland added since the last report. While capacity may be becoming less of an issue, there are still concerns that the certification process represents a significant bottleneck according to MedTech Europe with extended timelines, increased costs and uncertainty surrounding them, and lack of clarity around clinical evidence and documentation requirements all contributing.Additionally, just what “capacity” actually means in terms of ability of NB to certify devices may need better definition (Section 4).
Certification times ranging from 6-24 months with an average of 18 months mean that many companies are trying to get their devices registered in other jurisdictions first, or in parallel with the EU in order to bring their products to market sooner.
Again, these bottlenecks may disproportionately affect SMEs who likely have less experience navigating the regulatory landscape than do larger companies and also are less able to mitigate the effects of delays and increased costs.
Monitoring of Availability of Medical Devices on the EU Market
The Commission published the results of Monitoring of availability of medical devices on the EU Market - Study overview and 2nd survey results on 1st December 2025.Study data were gathered up to October 31st, 2024, and covered a number of aspects relating to the availability both of medical devices and IVDs. The full publication is available at: PowerPoint Presentation.
This summary is of the IVD section.
The study was performed using surveys and mainly focuses on medical devices; only about 16% of survey responses were about IVDs.
67% of the companies responding were medium, small or micro size (i.e. employing less than 250).
Up to 31st October 2024, there have been 1246 applications for IVD certification lodged with NBs of which 423 (34%) had had certificates issued.
In addition to the 823 outstanding applications, there are another 5362 applications for IVD previously regulated under IVDD and which are expected to be transitioned to IVDR.42% of manufacturers indicated that <50% of their products intended to be regulated under IVDR had been transferred. Around 20% of manufacturers said that they had less than 10% of their products transferred although this figure is down from the previous survey. On the plus side, 30% of manufacturers reported that they had 90%-100% of their products transferred to IVDR.
Regarding NBs, the survey results suggested that a number of manufacturers (37%) are still not engaging with NB to get their products regulated with the majority of those citing financial reasons and lack of clarity in the regulation for not proceeding. With 19 NB now operating, reasons other than NB capacity may be causing bottlenecks (Section 3), and those companies not engaging with NB may run into difficulties as the deadline for full compliance approaches. In this context, it’s interesting to note that the main reason for refusal of application cited by NB is “Insufficient notified body resources” suggesting that even once a company finds and engages with a NB, there may still be issues even if the application itself has no problems. Therefore, the issue of NB capacity may not be one of actual numbers of NB, but with functional capacity.
Despite this, numbers of certificates of conformity under IVDR are increasing, up to 423 covering 5419 devices (as catalogue numbers) as of 31st October 2024.Of these, 43% are Class C which covers companion diagnostics.
Time taken to prepare an application before submission to an NB was 6-12 months for most manufacturers although around a quarter of companies reported a preparation time of >19 months and a quarter reported <6 months, presumably a reflection of the familiarity of the manufacturer with the regulation and the requirements for their IVDs. Of concern is that Class C is over-represented in the groups with longer application preparation times. After submission, the most common time for certification is 13-18 months. Again, this may become an issue as the deadline for certification of legacy devices (i.e. those previously regulated under IVDD) approaches.
Another concern, repeatedly stated by stakeholders, is the regulatory burden imposed by IVDR compliance may lead to the withdrawal of some IVDs from the EU market. This has been recognised by the Commission (Section 2), perhaps as a result of the survey which reported that 40% of manufacturers had stopped supply of some IVDs into the EU market since 2022, and 37% planning to discontinue some IVDs. Of the different reasons given, 64% related to costs of compliance making the products economically unviable.
Laboratory-Developed Tests (LDT)
There is still no real clarity on implementation of IVDR in respect of LDT (also called in-house tests). Cobbaert and colleagues published an interesting opinion piece (Urgent call to the European Commission to simplify and contextualize IVDR Article 5.5 for tailored and precision diagnostics - PubMed) outlining some of the issues IVDR will cause to use of LDT and calling on the Commission to revise the regulation and take regard of the reasoning behind the recent US court decision to not allow the FDA to regulate LDT. Some of these changes have been put into the act amending MDR and IVDR (Section 2) but it’s clear that one of the main problems with the in-house exemption (Article 5(5)), that of LDT requiring justification for use if an identical test is placed on the market, will remain. The only concession the EU Commission gave on this was that there should be a “lengthy” transition period from a LDT to the commercially available product. The main reason labs use LDT is because they are generally cheaper than the corresponding commercially available test. The regulation would still have the effect of forcing labs to decide whether to use a more expensive test or to discontinue using their LDT. Also, as Cobbaert et al. point out, the regulation disincentivises labs to develop novel LDT if at some point a manufacturer launches a commercial version and the lab is obliged to use that instead of the test they developed. Oddly, it may also delay commercial development of novel tests if manufacturers wait to see what technology a non-commercial lab uses for a test, how it performs and what the demand is before making the decision to develop their own version.
Conclusions
It’s clear that progress on implementation of IVDR is being made. Long delayed, EUDAMED is now sufficiently in place for its use (at least for 4/6 modules) to become mandatory in May 2026.The Commission has recognised the “deluge of criticism” the regulation has received and is exploring ways to streamline and improve its implementation. There are now 19 NB.
However, issues remain.The availability survey suggested that functional capacity of NB may still be a problem, a number of IVD manufacturers have yet to engage with a NB while some are withdrawing products from the EU market, mainly due to economic considerations.There are still problems with the regulation around use of LDT.
The timescales for preparation of applications, review by NB and issue of certificates of conformity means that the deadlines for legacy devices (those previously regulated under IVDD) are in practical terms quite close: 2 years from now for Class D devices, 3 years for Class C and 4 years for Class B/sterile Class A.
It remains to be seen what stakeholders feel about the proposed revisions, how EUDAMED will perform under mandated (opposed to its current voluntary) load and how the problems surrounding use of LDT will play out.
IVDR Update: SEPTEMBER 2025
Is the EU Listening?
Our analysis of the results of the EU targeted evaluation of IVDR and MDR, plus news reports at the time, showed that most stakeholders were very dissatisfied both with the regulation and its implementation with some questioning the necessity for the regulation in the first place.
This has been confirmed by the release of the EU’s own summary report of the responses to the evaluation (Factual Summary Report on the Public Consultation for the Targeted Evaluation of the EU Rules on Medical Devices and In Vitro Diagnostics eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=PI_COM:Ares(2025)4926888)
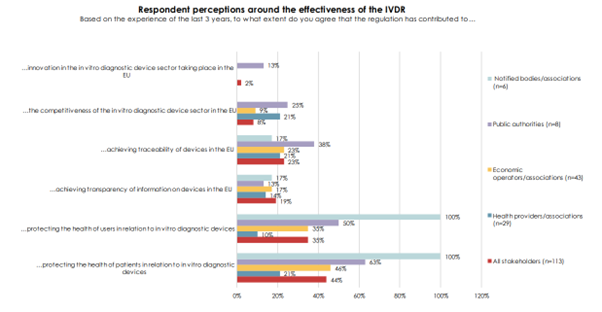
The chart shows that only 2% of all stakeholders think the regulation has contributed to innovation in the EU and only a minority think that the regulation improves the protection of IVD users and patients. Interestingly, around 80% of stakeholders felt that the regulation did not create a level playing field for all economic operators with less than 10% agreeing that costs of compliance with the regulation were acceptable. This is borne out by the experience of IVD manufacturers (see below).
There are signs that the EU is beginning to listen to the “torrent of criticism” it has received over the regulation with the recent (08SEP2025) publication of “Proposal for a Regulation of the European Parliament and of the Council amending Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices as regards simplification and burden reduction” (https://eur-lex.europa.eu/legal-content/AUTO/?uri=PI_COM:Ares(2025)7425764&qid=1757593574917&rid=1). It remains to be seen what comes of this initiative.
EUDAMED Still Lagging
The Commission’s phased rollout approach (enabled by Regulation (EU) 2024/1860) foresees “functionality” notices being published in the Official Journal of the EU (OJEU) for four modules first (Actors, UDI/Devices, NB & Certificates, Market Surveillance), with vigilance later. Publication, which was originally signpostedfor “around July 2025” is now generally referenced as “Q3 2025,” although no notice confirming module functionality has been published in OJEU.The EU did put a regulation into force in July 2025 enabling the gradual rollout of EUDAMED modules with a proposed timeline of Jan 2026 for mandatory use of the 4 functional modules and a timeline of Q2 2026 for mandatory use of the Vigilance module. However, the timeline is marked as “Under review, EUDAMED timeline July 2025) so it’s advisable to keep monitoring the situation and be prepared for compliance with the implementation.
Still Not Enough Notified Bodies?
There are currently 18 NB: 4 in Germany, 2 each in the Netherlands, Finland and Italy, and one each in Austria, Belgium, France, Ireland, Norway, Poland, Slovakia and Spain. This is still likely to be insufficient to meet demand for timely certification of devices (see below) and could lead to more problems with implementation of the regulation.
And More Guidance and Opinion
Medical Device Coordination Group (MDCG) has released guidance on IVDR testing (https://health.ec.europa.eu/latest-updates/mdcg-2025-5-questions-answers-regarding-performance-studies-vitro-diagnostic-medical-devices-under-2025-06-18_en) and software (https://health.ec.europa.eu/latest-updates/mdcg-2025-4-guidance-safe-making-available-medical-device-software-mdsw-apps-online-platforms-june-2025-06-16_en).
MedTech Europe has called for device sampling to be made more proportional to the risk classes of devices to avoid a disproportionate burden on in vitro diagnostic (IVD) manufacturers. MDCG guidance requires sampling of Class B and C devices under the IVD Regulation to be proportionate to the total number of devices under that certificate. MedTech Europe said “it is expected that 15% of technical documentation is reviewed per representative group of devices during each five years cycle which notified bodies may reduce to 5%.”Class B and C devices account for more than 90% of IVDs on the EU market, the trade group said, and as such the 15% sampling requirement “has a high burden on IVD manufacturers, especially small and medium-sized enterprises.” Average costs per one technical file assessment during an initial certification audit can total €38,000 ($43,000), according to MedTech Europe (Sampling under In Vitro Diagnostics Regulation: MedTech Europe proposal for a more risk-based approach - MedTech Europe).
Team-NB called for central governance, coordination and support for IVDR implementation. Team-NB, the European association of medtech notified bodies, published the paper with NBCG-Med, a notified body coordination group established as part of the medtech regulations. The groups framed their proposals as a response to a medical device regulatory framework that “faces fragmentation and inconsistent implementation, hindering safety, innovation and timely access.”
In response, the groups have proposed “a future governance model that integrates centralized support mechanisms, enhanced scientific and regulatory coordination and sustainable funding.” The plan is built around the Medical Device Coordination Office (MDCO), a proposed central coordination and support structure.
MDCO will oversee notified bodies, coordinate classification and qualification decisions, create guidance on how the legal system should work and develop technical and clinical guidelines. Other planned tasks for MDCO include coordinating consultation procedures, facilitating special pathways for certain devices and coordinating vigilance.
Notified bodies remain key players in the proposed system, with Team-NB and NBCG-Med making the case that their role as “as technical, regulatory and clinical ‘extended arm’ of the authorities should be maintained.”
“Moreover, notified bodies operate under strict designation and oversight criteria, ensuring independence, competence and impartiality. Their involvement in a centrally coordinated system — such as through the proposed MDCO — would enhance consistency and efficiency without compromising the decentralized strengths of the current model,” the notified body groups said.
The proposed model would give notified bodies advisory roles in decision-making structures. Team-NB and NBCG-Med said notified bodies could add value to expert committees, guidance development, special pathway procedures and classification discussions. The groups are also calling for NBCG-Med to get an expanded mandate to boost its contribution to guidance development and harmonization efforts.
Team-NB and NBCG-Med propose funding MDCO through a mix of user fees and contributions from the EU budget. Companies would pay “modest fees associated with activities in EUDAMED,” the groups said, and could pay additional fees for specific services such as early advice.
Another set of proposals covers special pathways and early advice. The notified bodies said they support the creation of a special pathway for devices that face disproportionate challenges under the medtech regulations, such as pediatric, orphan and breakthrough products.
“However, such pathways must not be implemented solely between manufacturers and individual notified bodies, as this could lead to inconsistencies and regulatory uncertainty. Instead, a coordinated process with clear guidance and oversight is essential,” the notified bodies said.
The notified bodies proposed adding a special pathways annex to the medtech regulations to define the eligibility criteria, minimum data requirements and other details. The proposals include a suggested process for seeking special pathway status. (NB-Perspective-on-Future-Governance-in-EU-medical-device-sector-20250728.pdf)
Webinar Encapsulates Manufacturers’ Problems
A recent webinar by 360DX highlighted some of the problems being faced by IVD manufacturers, particularly SMEs as they try to navigate compliance and launch devices onto the EU market.
The panel highlighted several problems with NBs:
NB aren’t allowed to consult so questions about issues with technical documentation can be very vague to avoid accusations of coaching. This leaves manufacturers trying to identify the problem with their documentation before they can begin to address it.
Manufacturers know that getting to grips with the detail of the IVDR entails a steep learning curve and this also applies to NB. One panellist in the webinar stated that “NB still need to find their feet” and this can lead to delays in review of documentation. One company switched NB after being told by their original NB that there would be a 9-month delay before examination of their submission.
The panellists had experienced challenges in terms of consistency of NBs with divergence of opinion in approach. Manufacturers don’t have access to all (e.g.) Team NB papers and may disagree with their positions.
It was agreed that the EU is still working out the process of implementation. Delays affect planning, cause hesitancy in responses from EU and may create a false sense of time for companies leading them to think they have longer to get things done than they actually have.
The panel also identified issues with guidance with one panellist describing the situation as “a lot of guidance but not enough guidance”. For context: IVDR 300 pages of regulations with the same quantity of implementation legislation. There are over 2000 pages of MDCG guidance. This makes it difficult for small QA/Reg teams to keep up with all relevant legislation giving them so much guidance it causes a bureaucratic overload and suggests the original legislation was not well written. In addition to this, most guidance relates to the MDR not IVDR, and IVDR guidance is delayed relative to MDR forcing companies to use MDR templates/guidance for IVD which are often not appropriate.
There was some debate over whether IVDR offers any actual benefits over the previous legislation, IVDD (see the section on the evaluation, above)
Some felt that it gave more confidence in safety of devices perhaps but that it may not provide more patient safety. Panellists felt that the Summary of Safety and Performance (SSP) does not provide any additional safety, so it becomes, in effect, just a bureaucratic exercise.
Some panellists thought that it could bring benefits but right now posed lots of challenges with uneven guidance and interpretation across countries. Harmonisation should be occurring but isn’t. One of the biggest problems is implementation started before all the infrastructure elements were put in place (see the section on EUDAMED, above).
Because of these difficulties, companies don’t know timeline and costs of placing a product onto the market and need improved transparency about costs and timeline.
The increased regulatory burden translates into increased costs but this is not reflected in reimbursement of test costs in many EU countries, which makes things especially difficult for SMEs.
These problems mean that many companies are choosing to launch new products in jurisdictions other than the EU, delaying access to new innovative tests which could have detrimental effects for patients. This likely underpins the evaluation finding that only 2% of respondents thought the regulation contributed to innovation in the EU (above).
Conclusion
Implementation of IVDR is still very much a work in progress although progress is being made, albeit at a slow pace. There are problems across the entire range of implementation activities for the regulation, and this is potentially restricting access to innovative new tests for the patients in the EU.
IVDR Update: JUNE 2025
Analysis of Feedback Given to EU on IVDR
Background
The EU Commission ran a public consultation exercise on the Medical Devices Regulation (MDR, (EU)2017/745) and the In Vitro Devices Regulation (IVDR, (EU)2017/746). Evaluation of the 2 regulations ran from 12th December 2024 to 21st March 2025.During this time, the Commission received 584 responses, 175 of which were related to IVDR (responses searched using the keyword “IVDR”).
Baseline data relating to demographics of respondents were extracted using word counting from the relevant form fields, however in view of the complexity of the free-text responses about the regulation, we used AI to perform quantitative analysis of the responses according to six separate themes derived from manual review of all the IVDR-related submissions. These were:
- Regulatory Burden
Multiple respondents emphasized the excessive complexity and costs of compliance with the IVDR. This is particularly challenging for small and medium-sized enterprises (SMEs), hindering their ability to bring innovative medical technologies to market. - Commercial Concerns
Concerns were raised about the costs of compliance, especially for companies who previously self-certified under the In Vitro Devices Directive and were, therefore, having to engage expensive consultancy because of their lack of familiarity with the certification process. - Impact on Laboratory-Developed Tests (LDTs)
Concerns were raised about the IVDR's effect on laboratories' ability to develop and use LDTs. This includes the challenges of meeting regulatory requirements, costs, and fear of losing in-house testing capabilities. - Stifling Innovation
The regulations are perceived to stifle innovation, particularly for SMEs and start-ups. Respondents argued that regulatory complexity discourages investment and delays the entry of new technologies into the EU market. - Clinical Trials and Market Competitiveness
Some feedback pointed out the impact IVDR could have on clinical trials for new drugs, causing delays and increasing costs. This, in turn, would affect the EU's attractiveness as a market for innovative products. - Governance and Practical Implementation Issues
Issues with consistency both of governance and the practical implementation of the regulations were mentioned, highlighting a need for better alignment and understanding across member states.
Results
Fig.1-3 show the proportions of the responses by country, user type and organisation size, respectively.
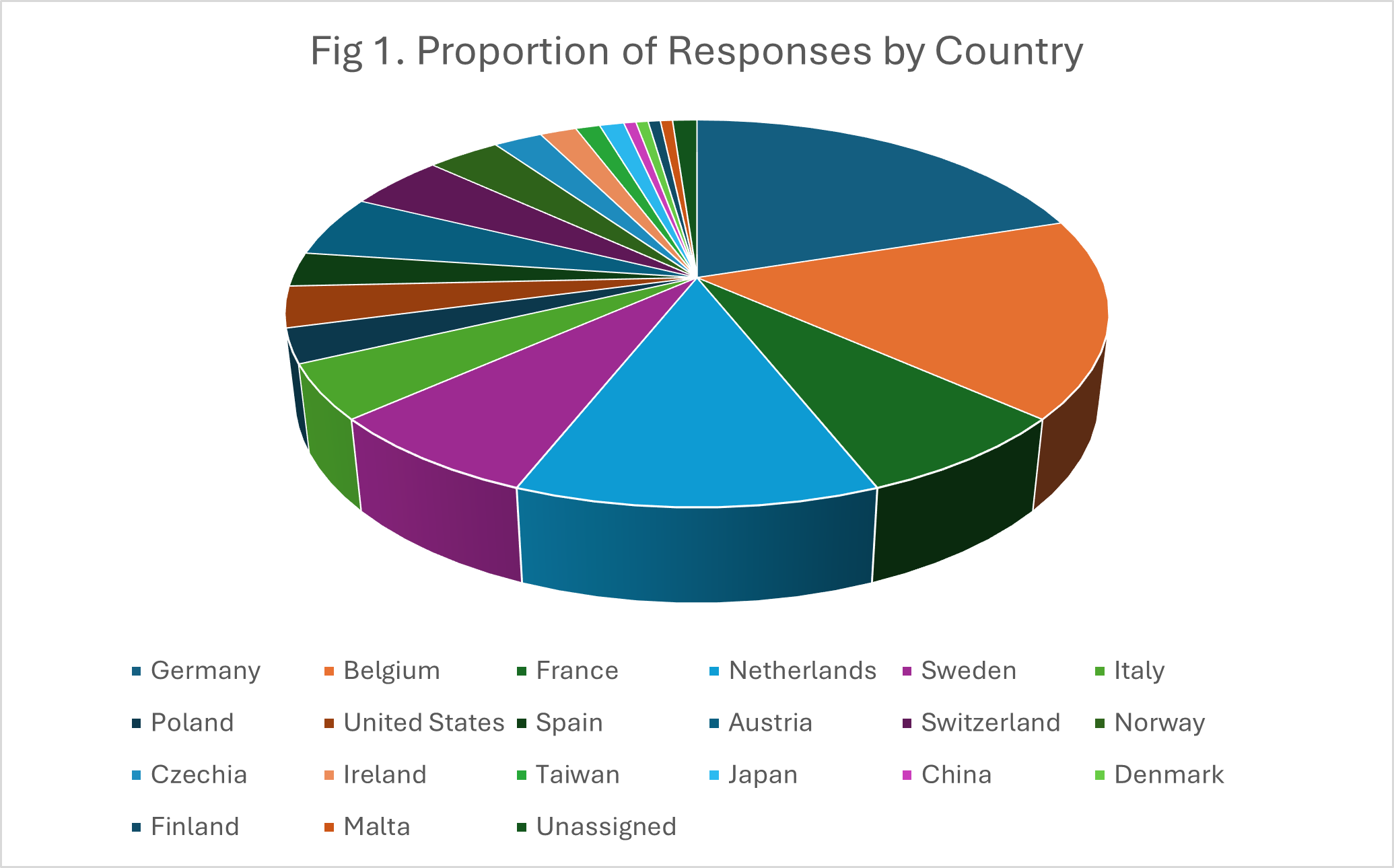
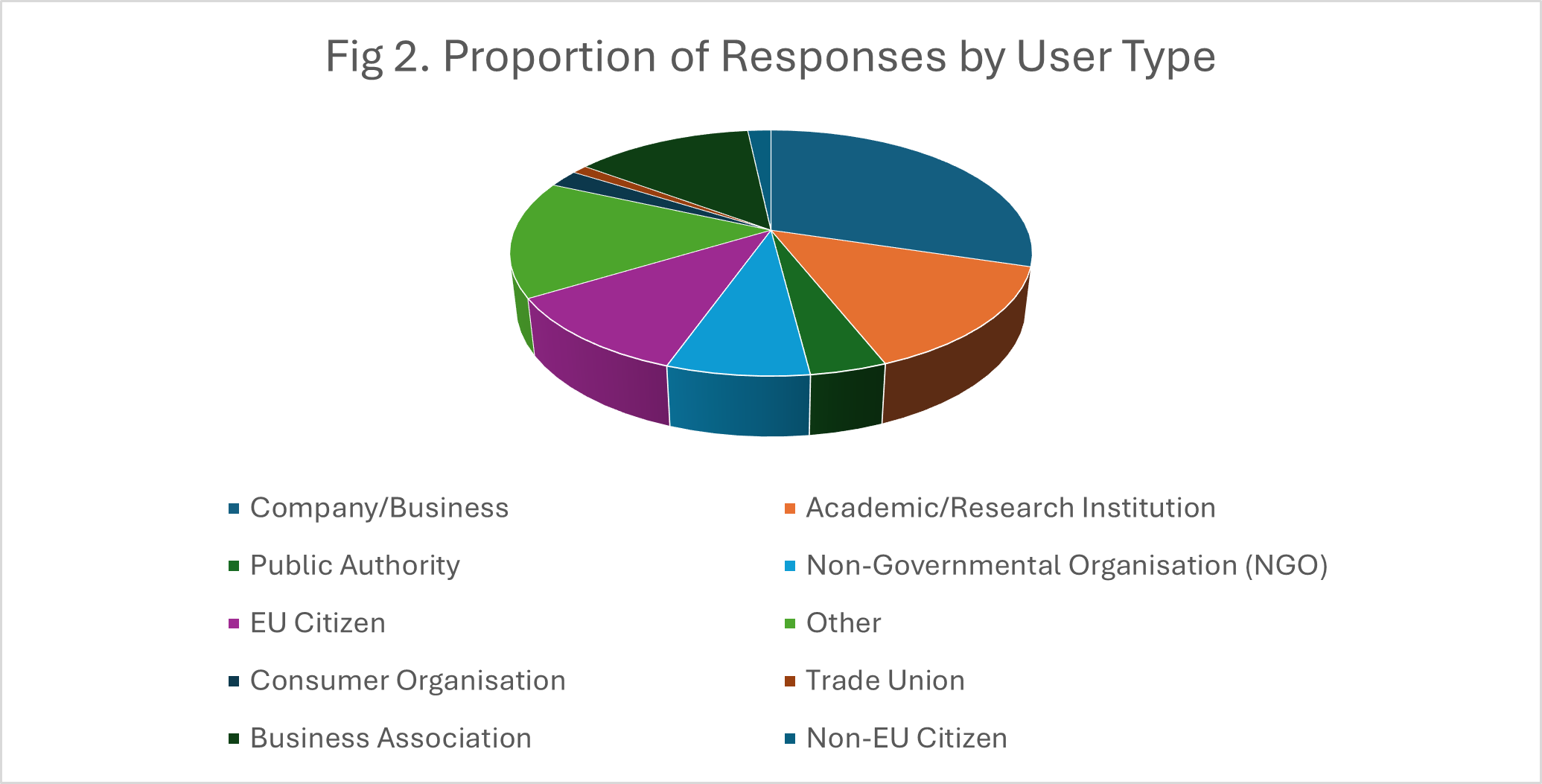
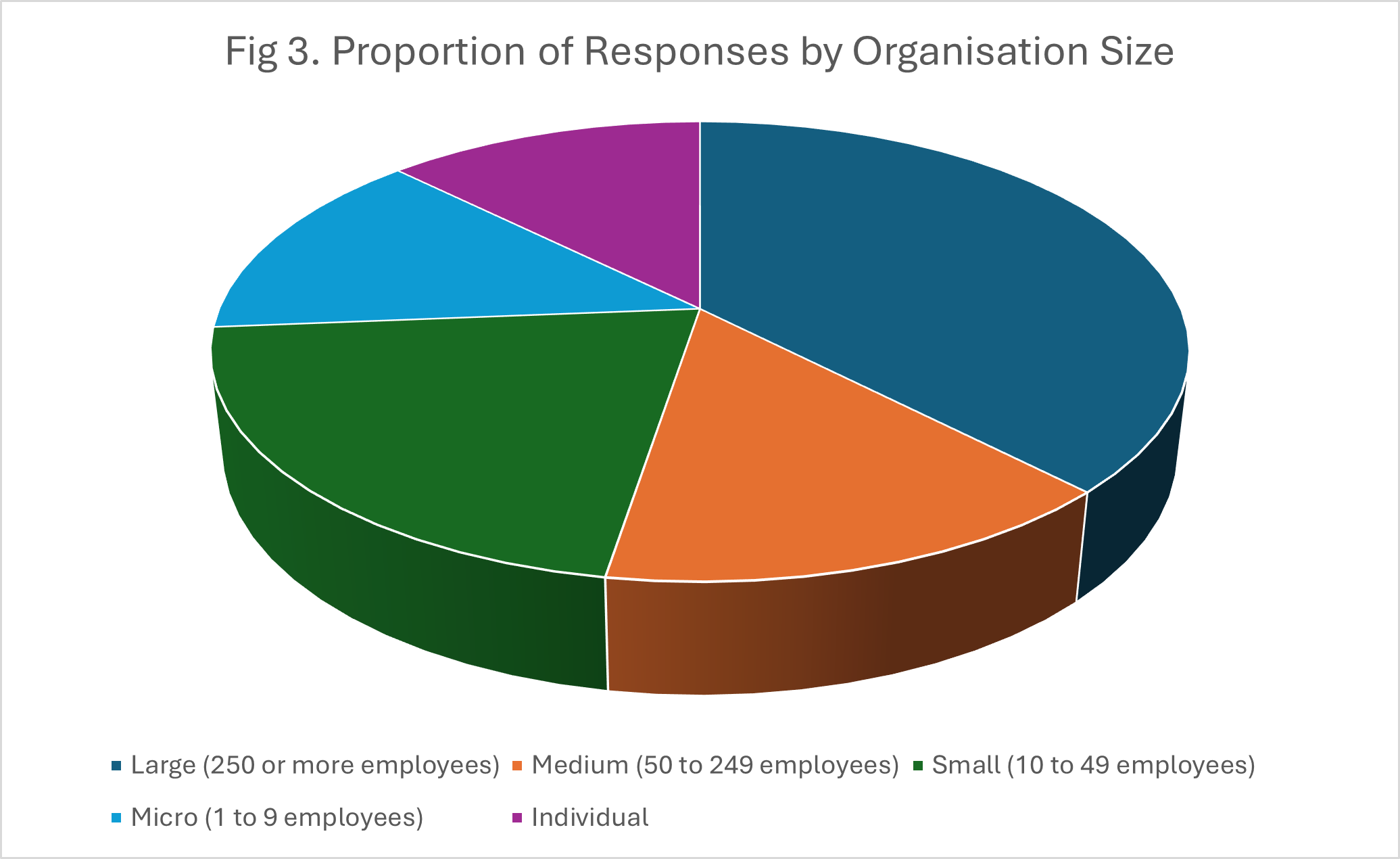
AI-based analysis mapped the numbers of responses to the areas given above with counts by particular sub-themes. These are given in Table 1.Categorisation across the main themes clearly produced overlapping response e.g. a major concern was that the burden imposed by regulation could affect many areas of IVD use including clinical trials, LDTs, commercialisation of existing devices and innovation/launch of new devices.
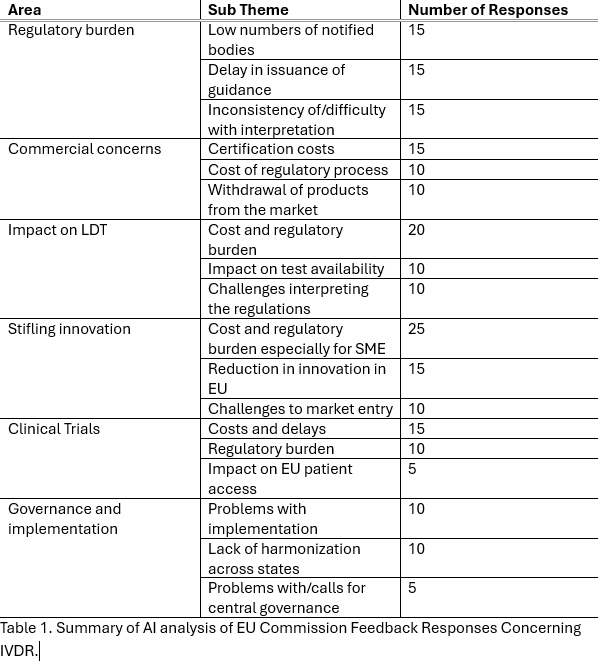
Conclusions
The Commission received hundreds of responses in its request for feedback about MDR and IVDR. An analysis of 175 of these responses relating to IVDR showed that respondents came from both across the EU and outside it, the latter presumably reflecting the concerns of those in non-EU countries trying to sell IVDs into the Union. A range of stakeholders were represented, from academic institutions and hospitals to commercial companies to individual citizens. The size of the stakeholder organizations was spread across a wide range, from individuals to large organizations/companies with up to several thousand employees.
Most respondents were highly critical of IVDR, many expressing concerns about the effect of the regulation on the availability to EU citizens of crucial medical tests, its adverse effects on clinical trials run in the Union and on innovation. In addition to this, many responses highlighted the costs and difficulty of regulatory compliance especially for small companies and non-commercial organizations such as hospitals and academic centres. Some questioned the need for there to be a new regulation at all opining that existing regulations such as ISO15189 were sufficient for laboratories. While some respondents agreed with the aim of the regulation, that of improving patient safety, many felt that this was compromised by issues with the regulation itself, the costs of compliance and problems with implementation such as the small number of Notified Bodies and lack of EUDAMED modules.
The Commission asked people what it thought of IVDR and received a “torrent of criticism” (Medtech Insight, EU Medical Device Regulations Face Torrent of Criticism In Public Consultation ) with comments like: “The IVDR is a single disaster…. Instead of improving patient care as originally requested by EU law, the exact opposite conclusion has been reached”. It will be interesting to see how it responds and what, if any, changes will be made to the regulations in the light of so many adverse comments.
IVDR Update: APRIL 2025
Still not enough Notified Bodies
Spain got its first Notified Body (NB) in February when the Centro Nacional de Certificatión de Productos Sanitarios (CNCps) received its designation. The Irish NB The National Standards Authority of Ireland (NSAI) had its designation temporarily suspended in December 2024 but was reinstated on 18 March 2025 after process improvements identified in an audit were put in place. There are, therefore, currently 14 NB listed as active and able to take on certification work. There is likely still not enough NB to provide conformity assessment to all devices that require it, especially given the time taken for the process. Estimates of the time taken for certification vary: one survey found that 75% of devices received certification in 13-18 months with 17% taking 19-24 months with the chief limitations being the tightened regulatory requirements and limited time and capacity of notified bodies. These problems are exacerbated by the fact that many manufacturers may be interacting with a NB for the first time and are unfamiliar with several of the processes involved. Also, in order to maintain impartiality, NB are not allowed to act as consultants to clients for whom they are undertaking conformity assessments – they can’t “coach” a client through the process.
NB may refuse or return applications and the most common reasons are: outside of the scope of the NB, wrong classification of the device, wrong conformity assessment procedure or incomplete application.
The structure and quality of the technical documentation supporting the application can also be problematic, again made worse by an applicant’s unfamiliarity with the process and the documentation required. Here, the role of consultancies with expertise in the field, but not acting as NB, may be vital to help manufacturers get the certification they need for their products but will likely add considerably to the costs and possibly be in short supply.
Certification costs vary considerably among NB with flat fees for application of the order of several thousand € and separate fees for certification audits and assessment of technical documentation running several hundred € per hour or several thousand € per day depending on the NB. Overall, certification costs could be a minimum of €50k with the potential to go much higher, an even higher if consultancy is required. The EU mandates that all NB publish their fees for the sake of transparency and this information is available HERE
So, a lot of the issues with NB are still present and it remains to be seen how many low margin products become economically unsustainable because of regulatory costs and are, therefore, pulled from the market.
EUDAMED – the plot thickens…or does it?
So far just 3 out of 6 EUDAMED modules are operational and that only on a voluntary basis. These are:
- Actor Registration: Information on manufacturers, importers, distributors, and other relevant parties.
- UDI/Device Registration: Details on unique device identifiers (UDIs), device classification, and registration.
- Notified Bodies and Certificates: Information about notified bodies and the certificates they issue.
The other 3 modules are:
- Vigilance: Reporting of adverse incidents and recalls.
- Market Surveillance: Oversight of medical devices on the market.
- Clinical Investigations and Performance Studies: Data on clinical trials and performance studies.
It is anticipated that use of the first 3 modules, and of the market surveillance module, will become mandatory some time in 2026 with the timeline for use of the remaining 2 modules uncertain but possibly late 2026 or 2027.
In addition to huge delays – EUDAMED was intended to be operational in 2021 – there have been allegations, somewhat contentious, that the costs related to development of the databases and systems have spiralled, that the project is over time, over budget and over staffed. Whether these allegations are unfounded or not, there are still many uncertainties over how, and on what timescale, EUDAMED will become fully operational.
Be careful what you ask for…
The EU Commission called for feedback on both the MDR and IVDR with a consultation period extending from 12th December 2024 to 21st March 2025 and adoption of “fine tuning” of the regulations due in Q4 2025.There are currently 398 responses and, according to Medtech Insight, the Commission has faced a “torrent of criticism” of the regulations. Recurrent themes in the feedback include the complexity of the regulations, the difficulty and cost of their implementation (see comments about consultancy, above) and their potential to stifle innovation.
Once the consultation is closed, we’ll analyse the feedback in terms of the problems labs are having with implementing the regulations for laboratory developed tests (LDT, also called the in-house exemption, Article 5.5 IVDR) and present this as a special review.
Discontinuation of supply
In December, the EU provided guidance on “The Information Obligation in Case of Interruption or Discontinuation of Supply of Certain Medical Devices and In Vitro Diagnostic Medical Devices”. As of 10th January 2025, manufacturers will have to inform the EU if they anticipate an interruption in the supply of a device or intend to discontinue it in the EU if the non-availability of the device carries a risk of serious harm to patients or public health in one or more member states of the EU. Completed notifications will have to be cascaded down through economic operators e.g. distributors to final customers. Notification must be provided at least six months prior to anticipated interruption or intended discontinuation.
Included in the guidance as the phrase “It should be noted that serious harm can also be due to the inability of a healthcare professional to deliver a specific medical treatment due to an interruption or discontinuation in the supply of a device.” (our emphasis).This may have special relevance in the case of companion diagnostics where the non-availability of a specific test may interfere with the use of the therapy which is gate-keepered by the companion diagnostic.
However, the regulation does not apply to “custom-made devices”. While not clear, this, and application of the regulation to manufacturers, suggests that labs who make LDTs covered by the in-house exemption may not have to comply with the notification requirements.
So, problems remain with NBs, EUDAMED is still problematic and well behind schedule and a lot of very unhappy people have told the EU what they think of the regulation.
Watch this space…